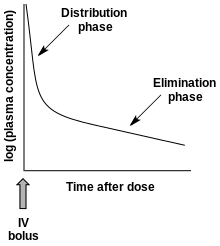
藥代動力學通過非膈室或膈室方法進行建模。非隔室方法可通過估算血藥濃度-時間圖中的曲線下的面積(Area under the cruve,AUC)估計藥物暴露量(Exposure)。隔室方法使用動力學模型估算血藥濃度-時間圖。非隔室方法不假設任何特定的隔室模型,而同樣可準確預測生物等效性,因此被更廣泛地使用。[4]藥物在生物體內如何發生轉化以及化合物最終的排泄過程,取決於許多相互關聯的藥代動力學因素。為了簡化這方面的研究,已經開發了許多功能模型進行預測。這些模型基於將生物體視為許多相關隔室,如將有機體視為只有一個同類隔室。這種單室模型先假定藥物的血漿濃度可真實反映藥物在血漿體液或組織液中的濃度,並且藥物的消除與生物體中藥物濃度成正比,即一級動力學[10]。
^ 1.01.11.2Gabrielsson J, Weiner D (2006) Pharmacokinetic and pharmacodynamic data analysis: concepts and applications, 4th edn. Swedish Pharmaceutical Press, Stockholm
^Knights K, Bryant B. Pharmacology for Health Professionals. Amsterdam: Elsevier. 2002. ISBN 0-7295-3664-5.
^ 4.04.1Levy, Gerhard, Milo Gibaldi, and William J. Jusko. "Multicompartment pharmacokinetic models and pharmacologic effects." Journal of pharmaceutical sciences 58.4 (1969): 422-424.
^Ruiz-Garcia A, Bermejo M, Moss A, Casabo VG. Pharmacokinetics in drug discovery. Journal of Pharmaceutical Sciences. February 2008, 97 (2): 654–90. PMID 17630642. doi:10.1002/jps.21009.
^Nestorov, Ivan A., et al. "Lumping of whole-body physiologically based pharmacokinetic models." Journal of pharmacokinetics and biopharmaceutics26.1 (1998): 21-46.
^Rescigno, Aldo. "Synthesis of a multicompartmented biological model." Biochimica et biophysica acta 37.3 (1960): 463-468.
^Biopharmaceutics and Pharmacokinetics Considerations Volume 1 in Advances in Pharmaceutical Product Development and Research 2021, Pages 195-277
^Handbook of Pharmacogenomics and Stratified Medicine 2014, Pages 341-364
^Benet LZ, Galeazzi RL (1979) Noncompartmental determination of the steady-state volume of distribution. J Pharm Sci 48:1071
^ 16.016.1Mould, D. R., and Richard Neil Upton. "Basic Concepts in Population Modeling, Simulation, and Model‐Based Drug Development—Part 2: Introduction to Pharmacokinetic Modeling Methods." CPT: pharmacometrics & systems pharmacology 2.4 (2013): 1-14.
^International Union of Biochemistry (now International Union of Biochemistry and Molecular Biology). Symbolism and terminology in enzyme kinetics. Recommendations 1981. Eur. J. Biochem. 1982, 128 (2–3): 281–291. doi:10.1111/j.1432-1033.1982.tb06963.x.
^Stroppolo ME, Falconi M, Caccuri AM, Desideri A. Superefficient enzymes. Cellular and Molecular Life Sciences. September 2001, 58 (10): 1451–1460. PMID 11693526. S2CID 24874575. doi:10.1007/PL00000788.
^Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS, Milo R. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry. May 2011, 50 (21): 4402–4410. PMID 21506553. doi:10.1021/bi2002289.
^Copeland RA. Why Enzymes as Drug Targets? Enzyme are Essential for Life. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists Second. John Wiley & Sons, Inc. March 2013: 1–23. ISBN 978-1-118-48813-3. doi:10.1002/9781118540398.ch1.
^When Is Kidney Removal Necessary? Dr. Hunter Wessells, Hunter Wessells Reviewed: August 27, 2007
^International Union of Biochemistry (now International Union of Biochemistry and Molecular Biology). Symbolism and terminology in enzyme kinetics. Recommendations 1981. Arch. Biochem. Biophys. 1983, 234 (2): 732–740. doi:10.1016/0003-9861(83)90262-X.
^Drug Bioavailability Gary Price Deven A. Patel In: StatPearls.Treasure Island(FL): StatPearls Publishing; 2023 Jan. PMID 32496732 Bookshelf ID: NBK557852 2022 Jun 23.
^Bioavailability: A Pharmaceutical Review 2011 J Novel Drug Deliv Tech
^Bioavailability L. Davidsson, S.A. Tanumihardjo, in Encyclopedia of Human Nutrition (Third Edition), 2013
^Dose-independent pharmacokinetics of ondansetron in rats: contribution of hepatic and intestinal first-pass effects to low bioavailability. Yang SH, Lee MG. Biopharm Drug Dispos. 2008 Oct;29(7):414-26.doi:10.1002/bdd.628.PMID 18697186
^ 34.034.1Michael E. Winter, Mary Anne Koda-Kimple, Lloyd Y. Young, Emilio Pol Yanguas Farmacocinética clínica básica Ediciones Díaz de Santos, 1994 pgs. 8–14 ISBN84-7978-147-5, 9788479781477 (in Spanish)
^Factors Affecting the Bioavailability of Chemicals
Mikko Nikinmaa, in An Introduction to Aquatic Toxicology, 2014
^Hsieh Y, Korfmacher WA. Increasing speed and throughput when using HPLC-MS/MS systems for drug metabolism and pharmacokinetic screening. Current Drug Metabolism. June 2006, 7 (5): 479–89. PMID 16787157. S2CID 13612670. doi:10.2174/138920006777697963.
^Covey TR, Lee ED, Henion JD. High-speed liquid chromatography/tandem mass spectrometry for the determination of drugs in biological samples. Analytical Chemistry. October 1986, 58 (12): 2453–60. PMID 3789400. doi:10.1021/ac00125a022.
^Covey TR, Crowther JB, Dewey EA, Henion JD. Thermospray liquid chromatography/mass spectrometry determination of drugs and their metabolites in biological fluids. Analytical Chemistry. February 1985, 57 (2): 474–81. PMID 3977076. doi:10.1021/ac50001a036.
^Li, Xue; Martinez-Lozano Sinues, Pablo; Dallmann, Robert; Bregy, Lukas; Hollmén, Maija; Proulx, Steven; Brown, Steven A.; Detmar, Michael; Kohler, Malcolm; Zenobi, Renato. Drug Pharmacokinetics Determined by Real-Time Analysis of Mouse Breath. Angewandte Chemie International Edition. 2015-06-26, 54 (27): 7815–7818. PMID 26015026. doi:10.1002/anie.201503312. hdl:20.500.11850/102558(英語).
^Sheiner LB, Rosenberg B, Marathe VV. Estimation of population characteristics of pharmacokinetic parameters from routine clinical data. Journal of Pharmacokinetics and Biopharmaceutics. October 1977, 5 (5): 445–79. PMID 925881. S2CID 28622472. doi:10.1007/BF01061728.
^Robert N, Wong GW, Wright JM. Effect of cyclosporine on blood pressure. Cochrane Database of Systematic Reviews. January 2010, (1): CD007893. PMID 20091657. doi:10.1002/14651858.CD007893.pub2.
^Rowland M, Tozer T (2010) Clinical pharmacokinetics and pharmacodynamics: concepts and applications, 4th edn. Lippincott Williams & Wilkins, Maryland
^Jager T, Albert C, Preuss TG, Ashauer R. General unified threshold model of survival--a toxicokinetic-toxicodynamic framework for ecotoxicology. Environmental Science & Technology. April 2011, 45 (7): 2529–40. Bibcode:2011EnST...45.2529J. PMID 21366215. doi:10.1021/es103092a.



















































![{\displaystyle B_{A}={\frac {[ABC]_{P}\cdot D_{IV}}{[ABC]_{IV}\cdot D_{P}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e0349b27d143a975c5e5c3eeaf83b49e8c7a3318)
![{\displaystyle {\mathit {B}}_{R}={\frac {[ABC]_{A}\cdot {\text{dose}}_{B}}{[ABC]_{B}\cdot {\text{dose}}_{A}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/634ea7e9316eeae72a9904dcaf1ccb2c43511aaf)










 . PMID 6615450. doi:10.1042/bj2130561.
. PMID 6615450. doi:10.1042/bj2130561.